The Cancer-Related Analysis of Variants Toolkit (CRAVAT) is
an evolving web-based suite of informatics tools for
genomic variant interpretation that includes:
- variant mapping (genome<->transcripts<->protein sequence<->protein structure)
- extensive integrated annotation of variants, genes, and proteins
- variant impact scoring, including joint prioritization of all nonsilent variants
- structural and mechanistic visualization.
Results from CRAVAT submissions are explored in an interactive,
user-friendly web-environment with dynamic filtering and
sorting designed to highlight the most informative variants and
genes in your study. We provide parallel, high-throughput
processing of studies that can include many sequenced samples
and millions of variants. CRAVAT jobs can be run on
our public web-portal or you can run your own local CRAVAT server
as a Docker container. Programmatic interfaces and links to
our visualization services enable easy integration with
other methods for omics analysis.
Read
our most recent publication.
CRAVAT citation:
- Masica DL, Douville C, Tokheim C, Bhattacharya R,
Kim R, Moad K, Ryan MC, Karchin R (2017)
CRAVAT 4: Cancer-Related Analysis of Variants Toolkit.
Cancer Res., 77(21):e35-e38
- Douville C, Carter H, Kim R, Niknafs N, Diekhans M,
Stenson PD, Cooper DN, Ryan M, Karchin R (2013).
CRAVAT: Cancer-Related Analysis of VAriants Toolkit
Bioinformatics, 29(5):647-648
CHASM citation:
- Wong WC, Kim D, Carter H, Diekhans M, Ryan M,
Karchin R (2011). CHASM and SNVBox: toolkit for detecting
biologically important single nucleotide mutations in
cancer Bioinformatics, 27(15):2147-2148.
- Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE,
Kinzler KW, Vogelstein B, Karchin R (2009)
Cancer-specific high-throughput annotation of somatic
mutations: computational prediction of driver missense
mutations Cancer Res, 69(16):6660-7.
VEST citation:
- Douville C, Christopher, Masica DL, Stenson PD,
Cooper DN, Gygax DM, Kim R, Ryan M, and Karchin R
(2015) Assessing the Pathogenicity of Insertion and
Deletion Variants with the Variant Effect Scoring Tool
(VEST-indel) Human Mutation, doi: 10.1002/humu.22911.
- Carter H, Douville C, Stenson P, Cooper D, Karchin R
(2013) Identifying Mendelian disease genes with the
Variant Effect Scoring Tool BMC Genomics, 14(Suppl 3):S3.
Jobs are submitted to the CRAVAT web server simply by providing
an input set of variants and selecting a few options for
the type of analysis you would like. It is best to create
a login so that you can access the My Jobs page and
use the CRAVAT Interactive Results Viewer.
CRAVAT has a queuing system, with two separate queues
for small and large jobs. This is done so that
small jobs are not held up behind longer-running jobs.
Multiple concurrent jobs are run in both the large and
small queues. Currently, small jobs are defined as
those with 25,000 or less variants. 25,000 variants
will take approximately 1 hour for any analysis
assuming a normal server load. When jobs are submitted,
you will receive a rough estimate of the processing time.
The large jobs queue is capable of handling submissions
with millions of variants.
You may input small sets of variants by cutting/pasting them
into the textbox of the Input panel. In most cases, you will
want to submit your variants by uploading a file. Files must
be plain text files (not compressed). All genomic variant
positions should be in GRCh38 (hg38) coordinates. If you have
GRCh37 (hg19) coordinates, select the hg19 input panel
checkbox to liftover the variants to GRCh38. The specific
formats we accept are described below.
CRAVAT supports
VCF format v4.0 and above
as an input
format. VCF files are commonly provided by sequencing
centers and various variant and somatic mutation calling
software packages. CRAVAT processing uses variant calls
and sample identifiers from the VCF file for analysis. If
your VCF file contains information on call quality,
zygosity, total alternative reads, and total overall
reads (Fill in exact optional VCF field identifiers here),
then this information will be included in your CRAVAT
results. Note that CRAVAT results may include multiple
lines of results for a given VCF input line so that lines
containing multiple variants and multiple samples can be
annotated in detail individually. Note: the ID field in
VCF format will become the UID field in CRAVAT format. If
a VCF line is split into multiple result lines,
the sample name or other distinguishing characteristics
will be added to ID.
CRAVAT also supports a very simple text input format with
six fields separated by tabs or spaces. Each row represents
a variant/sample pair and must begin with a unique identifier
or UID. The UID must be in alphanumeric format
(no punctuation or symbols allowed). The sixth field is
a sample identifier and can be omitted if it is not
relevant. Comment lines are allowed. All lines that start
with ">", "#", or "!" are ignored as comments.
- Genomic-coordinate format (separated by a tab or a space):
# UID / Chr. / Position / Strand / Ref. base / Alt. base / Sample ID (optional)
TR1 chr17 7674188 - G T TCGA-02-0231
TR2 chr10 121520166 - G A TCGA-02-3512
TR3 chr13 48459831 + C A TCGA-02-3532
TR4 chr7 116777451 + G T TCGA-02-1523
TR5 chr7 140753336 - T A TCGA-02-0023
TR6 chr17 39724745 + G T TCGA-02-0252
Ins1 chr17 39724745 + - T TCGA-02-0252
Del1 chr17 39724745 + A - TCGA-02-0252
CSub1 chr2 39644095 + ATGCT GA TCGA-02-0252
Position is in 1-based open coordinates. For insertions
and deletions, use "-" as the reference base for insertion
and "-" as the alternate base for deletion. In the above
example, Ins1 a "T" inserted between the 39724745th and
the 39724746th bases. Del1 is an "A" at the 39724745th
position that is deleted. CSub1 is a complex substitution
in which "ATGCT" from the 39644095th to the 39644099th
positions is replaced by "GA". If you do not have strand
information from your sequencing results, it is likely
that they are all reported on the + strand. Make sure
that your reported reference base matches the base
in the reported position in the hg38 reference sequence
(or hg19 if you checked hg19 checkbox).
* The old format for indels, in which the base is
specified before the insertion/deletion location is
still supported. However, if this old format is used
in any row of your input, your entire input will be
handled in the old format. The old and new formats
cannot be mixed.
- HGVS format
CRAVAT also supports HGVS format for input of
variants. Currently we accept HGVS Missense (single
point) variants and insertions. The HGVS input includes
three tab delimited fields: 1. an optional identifier,
2. HGVS format variants, and 3. an optional sample
identifier. For example:
Var1 NC_000022.10:g.30025797A>T Sample1
Var2 NC_000022.10:g.40418496T>C Sample1
Var3 NC_000022.10:g.40419252C>T Sample1
Var4 NC_000002.10:g.218270043_218270044insG Sample1
- Transcript format
Missense variants can be entered in transcript format
# UID / Transcript / AA change / Sample ID (optional)
TR1 NM_001126116.1 D127Y TCGA-02-0231
TR2 NM_001144919.1 R162Q TCGA-02-3512
TR3 NM_000321.2 Q702K TCGA-02-3532
TR4 NM_000245.2 A1108S TCGA-02-1523
TR5 NM_004333.4 V600E TCGA-02-0023
The transcript identifier can be from either NCBI Refseq
(NM accessions), CCDS, or Ensembl (ENST accessions). Refseq
and CCDS accessions can be specified without version numbers.
CRAVAT is primarily designed for genomic input, and
submissions in transcript format are supported but
produce a limited set of annotations. Submitting variants in
genomic format is recommended.
By default, CRAVAT provides variant mapping across
genome<->transcripts<->protein sequence<->protein
structure, and extensive annotations (link to annotaions
documentation below). You can also select results of variant
and gene-level scoring algorithms (currently from
CHASM v3.1
and
VEST v4.0
and/or additional annotations from
GeneCards
and
PubMed.
CHASM (currently v3.1) provides cancer-specific missense
variant scores. If CHASM is selected, a list box that allows
you to choose a cancer type appears. VEST (currently v4) provides
pathogenicity scores for all non-silent variants.
Enter your email address (if you have logged in you don't
need to), and if you want to receive a machine
processing-friendly, tab-separated text version of the
CRAVAT analysis report in addition to its default Microsoft
Excel version, check "Include text reports for machine processing".
Then, click "SUBMIT". When all the analyses are complete,
an email with reports will be sent to you. If you have
logged in you can check the status and history of your jobs
at 'My Jobs' page.
If you have created a CRAVAT user account, the CRAVAT
server will track your job submissions and provide both
downloadable results and access to an interactive results
viewer. When logged in to your account, you can see
the status of your jobs and retrieve the results of
current or past jobs through "My Jobs" page.
Create a CRAVAT Account:
There are two ways to create a CRAVAT account:
-
When you submit a job for the first time, CRAVAT will create
an account with your email and a temporary password and
this account information will be sent to you as a part of
the result notification email.
-
A CRAVAT account can be created by clicking
"Log-In" > "Create an account" on the top menu.
Retrieve Your Username
Your username is your email.
Retrieve Your Password
When you create your login, you will setup a challenge
question and answer. If you forgot your password,
click "Log-In" > "Forgot password?". You will then enter
your login (your email) to retrieve your challenge
question. Answering your challenge question will reset
your password. The new password is displayed below
the challenge answer. Use the new password to login
and then you can reset your password.
Change Your Password
To change your password, first log in, and then click
"My Profile" > "Change password". In the "Change Password"
pop-up window, type your current password, your new
password, and again your new password. Click "Submit".
My Jobs Page
After having logged in, click "My Jobs" on the top menu
to open the My Jobs page in a new browser tab. This page
shows statistics in tabular and graphic format for your
past and current jobs and the status of each job
(success, fail, running, and in-queue). By clicking
"Download" in the "Result" column, you can download
Excel and/or text result files and by clicking "Explore"
you get access to the interactive interface (recommended).
Log in to the CRAVAT main page and click "My Jobs". This
will open a new browser tab with the following table
which shows your submitted jobs.
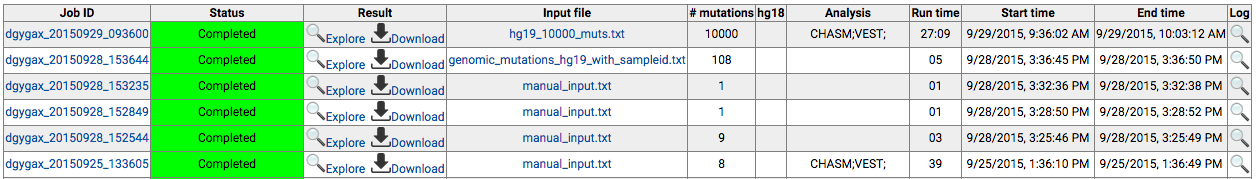
For completed jobs, if the interactive result viewer is
available, the "Explore" icon will show up, and if
result files are available for download, the "Download"
icon will show up. Clicking "Explore" will open a new tab
with the interactive result viewer. Clicking "Download" will
start downloading the results files.
The interactive result viewer contains five tabs:
Summary, Gene, Variant, Noncoding, and Error.
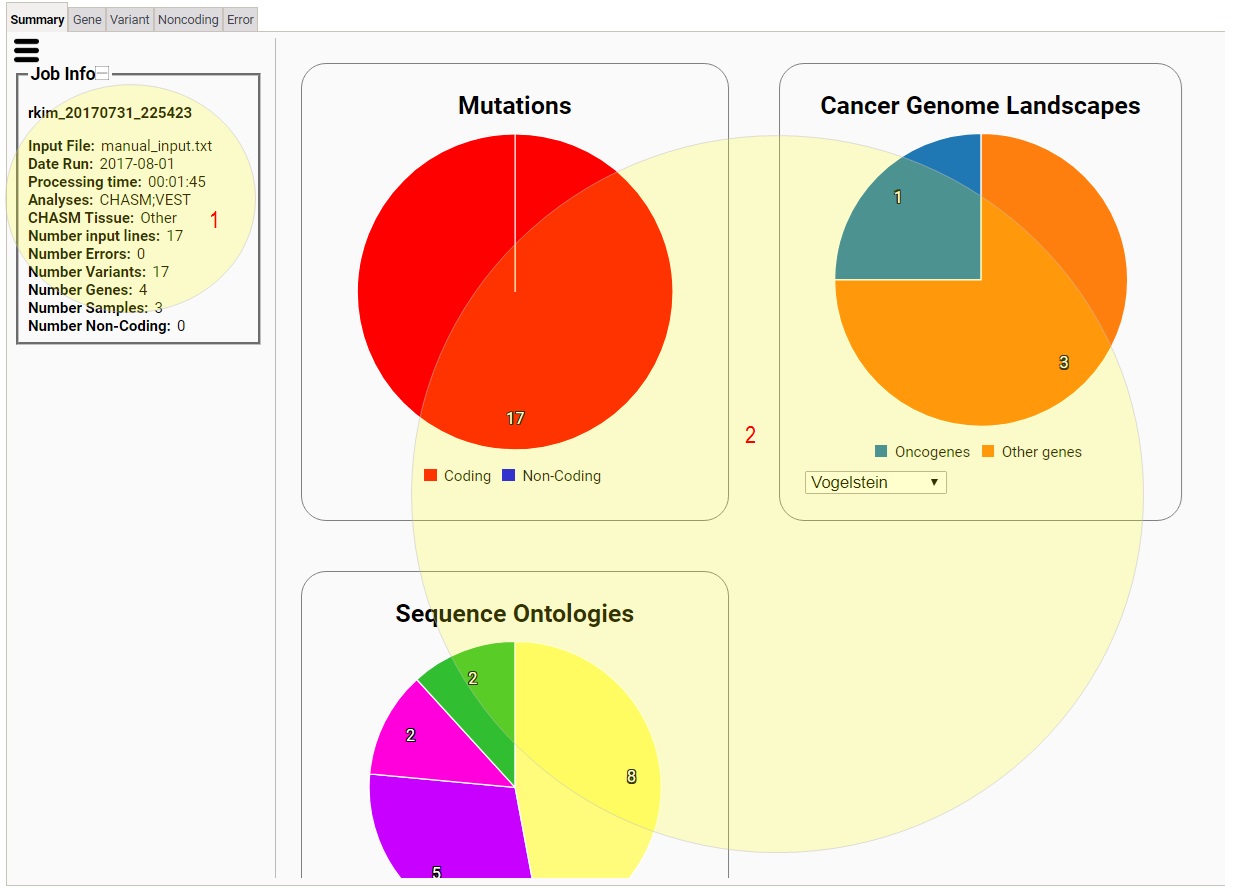
1) This tells you various information of your job.
2) This area has various widgets that show you summarized
statistics on your variants, which include breakdowns of
your variants according to coding/noncoding,
oncogene/tumor suppressors, sequence ontologies, and
sequence ontology and sample,
as well as a circular plot of human chromosomes on which
your variants are shown (Circos plot), top genes according
to mutation rate, CHASM scores, and VEST scores, and
Network Data Exchange (NDEX) network hits of your
variants.
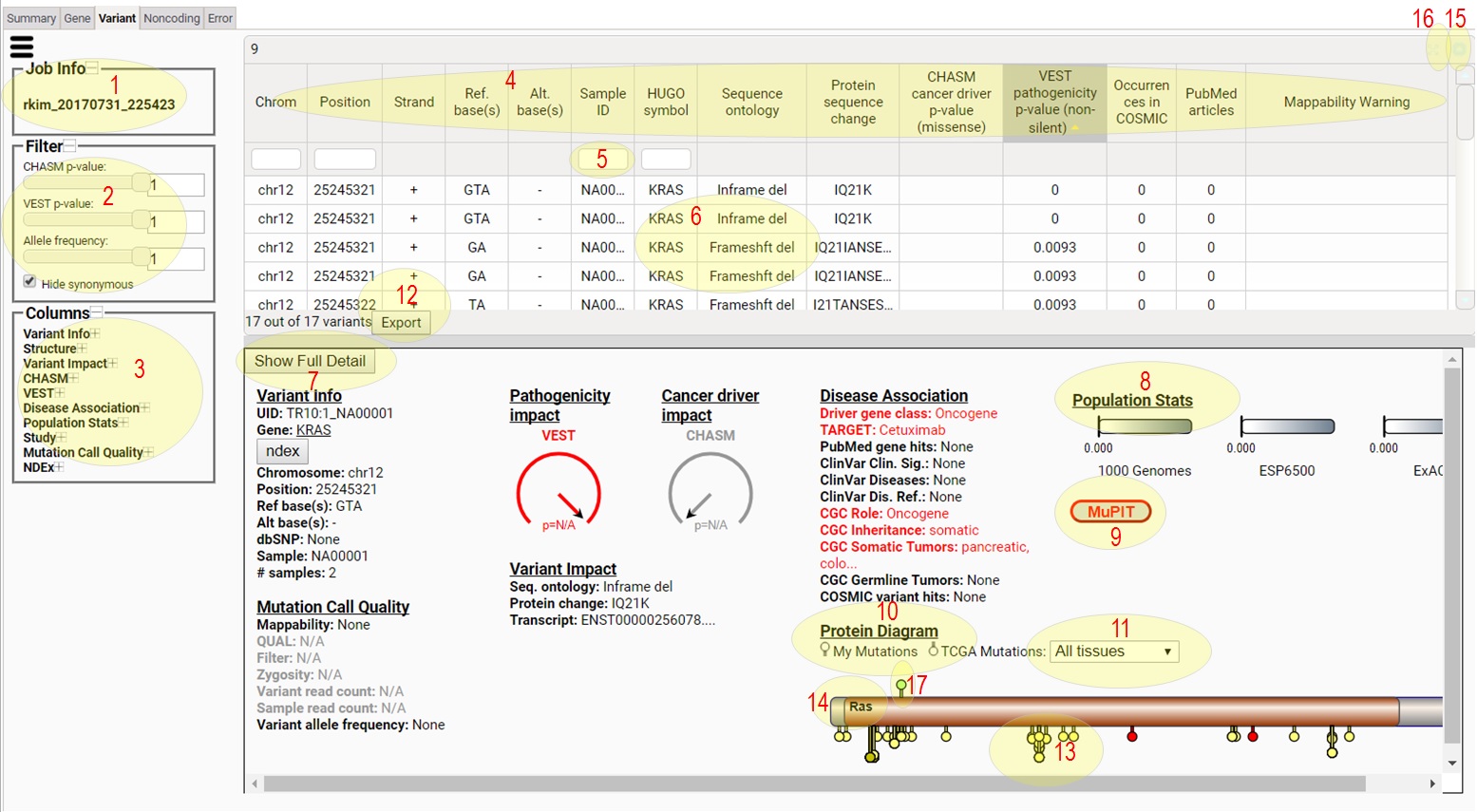
1) This tells you the Job Id for the CRAVAT job being viewed
in the Interactive Result Viewer.
2) The filter section allows you to change which variants
are shown in the variants table. You can move sliders to
set the values. You can click 'Hide synonymous' on or off
to show or hide synonymous variants. As you change filter
conditions, the table on the right will be updated
automatically.
3) The columns section can be used to turn on and off
the columns show in the variants table. Click the + and -
to expand or collapse column groups. Click on the column
names to show (dark grey) or hide (light grey) them.
4) Click a column header to sort the table. Shift-clicking
columns will do multiple-column sorting.
5) Type in column header search boxes to filter the table
for the rows that contain the typed text.
6) Click a gene on the gene table to the left and the
variants in study for the gene will show in this table.
7) Click to see
Network Data Exchange (NDEX) networks containing
the gene
8) If the selected variant or the variants in the selected
gene map to any PDB structure or high-quality model
structure, this button turns red. Click the red button
to see the structural mapping of the variant(s).
9) CHASM and VEST score and p-values are shown like
speedometers. The hand shows the score from 0 to 1,
and p-values are shown as "p=".
10) Protein domains, the selected variant(s), and the variants
from TCGA are shown as a lollipop diagram.
11) Use this to change the tissue for TCGA mutations.
8) Allele frequencies from 1000 Genomes, ESP6500, and gnomAD
are shown as bar-meters.
12) Click to download the table content as a tsv file.
13) Known variants from TCGA are shown below the gene
bar. TCGA variants that have multiple samples are
taller.
14) Protein domains are shown as brown bars. Regions of
interest in the protein sequence are shown as red lines
or X's below the gene bar.
15) Click to minimize/restore the table.
16) Click to maximize/restore the table.
17) The variant(s) from the study for the gene is shown above
the bar.
| Color |
Sequence ontology of variant |
 |
Synonymous mutation |
 |
Missense mutation |
 |
Inframe indel |
 |
Frameshift indel |
 |
Splice site mutation |
 |
Stop loss mutation |
 |
Stop gain mutation |
 |
Complex substitution mutation |
1) This tells you the Job Id for the CRAVAT job being viewed
in the Interactive Result Viewer.
2) The filter section allows you to change which variants
are shown in the variants table. You can move sliders to
set the values. You can click 'Hide synonymous' on or off
to show or hide synonymous variants. As you change filter
conditions, the table on the right will be updated
automatically.
3) The columns section can be used to turn on and off
the columns show in the variants table. Click the + and -
to expand or collapse column groups. Click on the column
names to show (dark grey) or hide (light grey) them.
4) Click a column header to sort the table. Shift-clicking
columns will do multiple-column sorting.
5) Type in column header search boxes to filter the table
for the rows that contain the typed text.
6) This is the table area which shows the analysis result
for the chosen tab and filter conditions.
7) Click to change the level of detail (summary or full).
8) Allele frequencies from 1000 Genomes, ESP6500, and gnomAD
are shown as bar-meters.
9) If the selected variant or the variants in the selected
gene map to any PDB structure or high-quality model
structure, this button turns red. Click the red button
to see the structural mapping of the variant(s).
10) Protein domains, the selected variant(s), and the variants
from TCGA are shown as a lollipop diagram.
11) Use this to change the tissue for TCGA mutations.
12) Click to download the table content as a tsv file.
13) Known variants from TCGA are shown below the gene
bar. TCGA variants that have multiple samples are
taller.
14) Protein domains are shown as brown bars. Regions of
interest in the protein sequence are shown as red lines
or X's below the gene bar.
15) Click to minimize/restore the table.
16) Click to maximize/restore the table.
17) The variant(s) from the study for the gene is shown above
the bar.
| Color |
Sequence ontology of variant |
|
Synonymous mutation |
|
Missense mutation |
|
Inframe indel |
|
Frameshift indel |
|
Splice site mutation |
|
Stop loss mutation |
|
Stop gain mutation |
|
Complex substitution mutation |
Non-coding tab contains a subset of the elements of the
variant tab. Please see the explanation on Variant Tab.
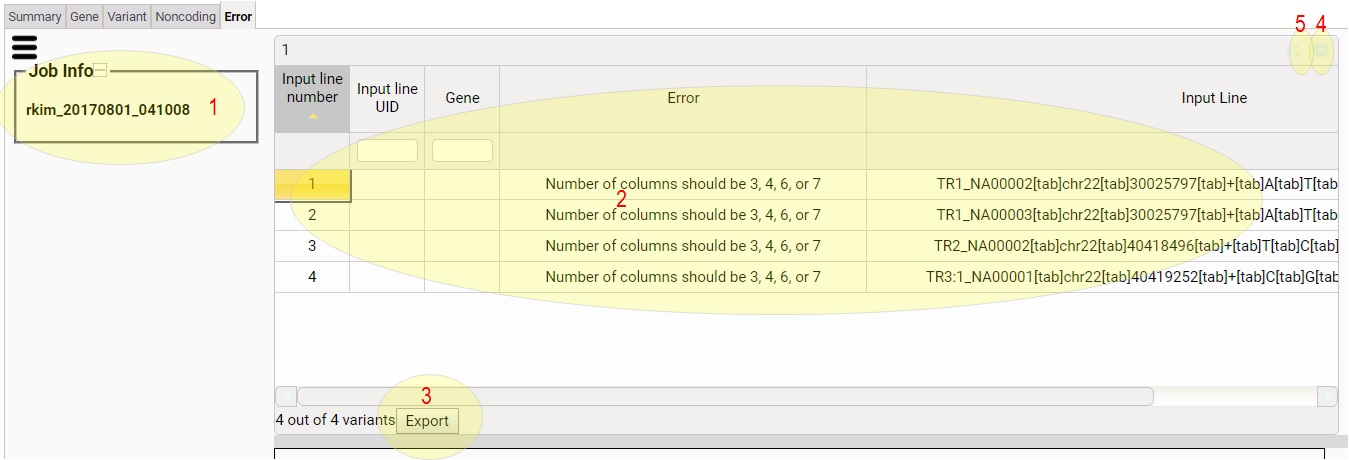
1) This tells you the job ID.
2) Error column shows the reason of each error input line. Input
Line column shows the input line
3) Click to download the table content as a tsv file.
4) Click to minimize/restore the table.
5) Click to maximize/restore the table.
Upon a successful submission and analysis, you will receive a link
to your results via email (if you have logged in you can check
the status and history of your jobs at 'My Jobs' page,
where you can also download your result by clicking
'Download' in the 'Result' column), which will be available
for 30 days from the date of submission. The results will be
delivered as one zip-compressed file containing several
report files, including a MS Excel format spreadsheet and
optional tab-separated text files. Five reports are included:
Variant, Variant Additional Details, Variant non-coding,
Gene-level analysis, and Input Errors. The spreadsheet has
a tab for each report, and the tab-separated text files have
each report a separate .tsv file. All reports are in table
format. The Excel spreadsheet file is provided only when
65,000 or fewer variants are analyzed.
There are two Galaxy Tools for querying CRAVAT.
To directly obtain annotations for your variants, use
https://toolshed.g2.bx.psu.edu/view/in_silico/cravat_annotate_mutations/1b6e23f3cb06.
To submit a job with your variants to benefit the full CRAVAT
analysis environment including its Interactive Result Browser, use
https://toolshed.g2.bx.psu.edu/view/in_silico/cravat_score_and_annotate/cdd97b06c802.
CRAVAT input format is used.
With CRAVAT's web service, you can submit and check
the status of your jobs without using a browser.
-
Job submission via POST
URL: http://www.cravat.us/CRAVAT/rest/service/submit
Method: POST
Consumes: Multipart/form-data
Produces: a JSON object, notable fields of which are as follows.
-
status: "submitted" for successful job submission,
"submissonfailed" for an error in the job submission
-
errormsg: If there was any error during the job submission,
the error message is written here.
-
jobid: The Job ID of the submitted job. This job ID can be
used to check the status of the job later using "status"
method which is explained below.
Form data parameters (* = essential parameters):
-
analyses: "CHASM", "VEST", "CHASM;VEST"
-
chasmclassifier: classifier name for CHASM analysis
-
*email: email of the submitter
-
functionalannotation: "on" or "off". GeneCards and PubMed
annotation.
-
hg19: "on" or "off". Input mutations are in hg19 coordinates or not.
-
*inputfile: Input mutation file. This is from the file input
element in the POST form.
-
mupitinput: "on" or "off". MuPIT input format returned or not.
-
tsvreport: "on" or "off". Text format reports returned or not.
Python example
>import requests
>r=requests.post('http://www.cravat.us/CRAVAT/rest/service/submit',
files={'inputfile':open('input_file/vcf_input.txt')},
data={'email':'test@test.com','analyses':'CHASM'})
>r.text # contains the submission result as a string. Check "status" field.
-
Job submission via GET
URL: http://www.cravat.us/CRAVAT/rest/service/submit
Method: GET
Produces: a JSON object, notable fields of which are as follows.
-
status: "submitted" for successful job submission,
"submissonfailed" for an error in the job submission
-
errormsg: If there was any error during the job submission,
the error message is written here.
-
jobid: The Job ID of the submitted job. This job ID can be
used to check the status of the job later using "status"
method which is explained below.
Query parameters (* = essential parameters):
-
analyses: "CHASM", "VEST", "CHASM;VEST"
-
chasmclassifier: classifier name for CHASM analysis
-
*email: email of the submitter
-
functionalannotation: "on" or "off". GeneCards and PubMed
annotation.
-
hg19: "on" or "off". Input mutations are in hg19 coordinates
or not.
-
*mutations: a string with mutations, the format of which is
the same as described in the "Input" section above.
-
mupitinput: "on" or "off". MuPIT input format returned or not.
-
tsvreport: "on" or "off". Text format reports returned or not.
Python example
>import requests
>r=requests.get('http://www.cravat.us/CRAVAT/rest/service/submit',
params={'email':'test@test.com', 'analyses':'',
'mutations':'TR1 chr22 30025797 + A T sample_1'})
>r.text # contains the submission result as a string. Check "status" field.
-
Job status checking
URL: http://www.cravat.us/CRAVAT/rest/service/status
Method: GET
Produces: a JSON object, notable fields of which are as follows.
-
status: "running" for still running, "success" for successful
completion, "jobfailed" for failed
-
errormsg: Error message if the job failed.
-
resultfileurl: If the job completed successfully, the URL of the
result file.
Query parameters (* = essential parameters):
-
*jobid: The job ID to query.
Example
http://www.cravat.us/CRAVAT/rest/service/status?jobid=test_20140204_102423
Python example
>import requests
>r=requests.get('http://www.cravat.us/CRAVAT/rest/service/status',
params={'jobid':'test_20170315_103245'})
>r.text # contains the job status as a string.
-
Single variant web API
URL:http://www.cravat.us/CRAVAT/rest/service/query
Method: GET
Produces: a JSON object, notable fields of which are as follows.
See Annotation Description for explanation on
the returned fields.
Query parameters (* = essential parameters):
-
*mutation: The chromsome, position, strand direction, reference
base and alternate base of the variant separated by underscores
(chomosome_position_strand_refBase_altBase)
Example
http://www.cravat.us/CRAVAT/rest/service/query?mutation=chr22_40418496_-_A_G
Python example
>import requests
>r=requests.get('http://www.cravat.us/CRAVAT/rest/service/query',
params={'mutation':'chr22_30025797_+_A_T'}) # query mutation is of the format chromosome_position_strand_reference_alternate.
>r.text # contains the annotation for the query mutation.
It is possible to display the details of a single variant as a full page on
a browser window. Use the following URL scheme to open a
Single Variant Page for the variant encoded in the URL.
CHASM-3.1 is the most recent version of "Cancer-specific
High-throughput Annotation of Somatic Mutations" a method
that predicts the functional significance of somatic missense
mutations observed in the genomes of cancer cells, allowing
mutations to be prioritized in subsequent functional studies,
based on the probability that they give the cells a selective
survival advantage.
Original CHASM publication
CHASM-3 overview
CHASM-3.1 was trained on
SNVBox (v5.0)
(updated and rebuilt for GRCh38) with
driver training examples
previously used in CHASM-3 and passenger training examples
generated according to dinucleotide frequencies in
25 Cancer Genome Atlas cancer types.
Cancer-specific classifiers
|
Name
|
Full name
|
|
Bladder
|
Bladder Urothelial Carcinoma
|
|
Blood-Lymphocyte
|
Chronic Lymphocytic Leukemia
|
|
Blood-Myeloid
|
Acute Myeloid Leukemia
|
|
Brain-Glioblastoma-Multiforme
|
Glioblastoma Multiforme
|
|
Brain-Lower-Grade-Glioma
|
Brain Lower Grade Glioma
|
|
Breast
|
Breast Invasive Carcinoma
|
|
Cervix
|
Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma
|
|
Colon
|
Colon Adenocarcinoma
|
|
Head and Neck
|
Head and Neck Squamous Cell Carcinoma
|
|
Kidney-Chromophobe
|
Kidney Chromophobe
|
|
Kidney-Clear-Cell
|
Kidney Renal Clear Cell Carcinoma
|
|
Kidney-Papillary-Cell
|
Kidney Renal Papillary Cell Carcinoma
|
|
Liver-Nonviral
|
Hepatocellular Carcinoma (Secondary to Alcohol and Adiposity)
|
|
Liver-Viral
|
Hepatocellular Carcinoma (Viral)
|
|
Lung-Adenocarcinoma
|
Lung Adenocarcinoma
|
|
Lung-Squamous Cell
|
Lung Squamous Cell Carcinoma
|
|
Other
|
General purpose
|
|
Ovary
|
Ovarian Serous Cystadenocarcinoma
|
|
Pancreas
|
Pancreatic Cancer
|
|
|
|
Prostate-Adenocarcinoma
|
Prostate Adenocarcinoma
|
|
Rectum
|
Rectum Adenocarcinoma
|
|
Skin
|
Skin Cutaneous Melanoma
|
|
Stomach
|
Stomach Adenocarcinoma
|
|
Thyroid
|
Thyroid Carcinoma
|
|
Uterus
|
Uterine Corpus Endometriod Carcinoma
|
VEST-4 is the most recent version of the Variant Effect
Scoring Tool. VEST is a machine learning method that
predicts the functional significance of non-silent
variants based on the probability that they are pathogenic.
Original VEST missense publication.
Original VEST insertion/deletion publication.
Changes from VEST3 to VEST-4
-
SNVBox features were updated and rebuilt for GRCh38 (SNVBox5.0)
-
Positive class expanded and updated to HGMD (2017.1)
-
Neutral class changed to ExAC Release 1 (2/2017)
-
Improved p-value calculations using non-training set negatives from ExAc and ESP6500 to seed a Gibbs Sampler algorithm. This technique produced a table of p-values with increased precision for all VEST scores for each sequence ontology.
| Field |
Description |
| Chromosome |
Chromosome |
| Position |
Genomic position in chromosomal coordinates |
| Strand |
Positive or negative |
| Reference base(s) |
Base(s) at position in the reference genome (hg38) |
|
Alternate base(s) |
Alternate base(s) |
| Sample ID |
Alphanumeric identifier of sample |
| S.O. all transcripts |
Sequence ontology for each transcript the variant is
mapped to. * = transcript with most severe sequence
ontology. |
| S.O. transcript |
Transcript with most severe sequence ontology. To break
ties, the longer transcript is chosen. |
| S.O. transcript strand |
The strand (+ or -) of the transcript used to assign
sequence ontology |
| Protein sequence change |
Protein sequence change produced by the variant. |
| Phred |
Phred-scaled quality score Available only with
VCF input. |
| VCF filters |
Status of VCF filters. PASS if the all are satisfied. Otherwise,
a semicolon-separated list of codes for filters that fail
(e.g. "q10;s50"). Available only with VCF input. |
| Zygosity |
Homozygous or heterozygous status of the variant. Available
only with VCF input. |
| Alternate reads |
Count of reads with alternate allele aligned to the
position. Available only with VCF input. |
| Total reads | Count of all reads aligned to the position. Available only with VCF input. |
| Variant allele frequency | Alternate reads / Total reads |
| ClinVar | ClinVar annotation of pathogenicity. Only the variants with "pathogenic" clinincal significance are reported. |
| dbSNP | dbSNP identifier |
| 1000 Genomes AF | Healthy population allele frequency from the 1000 Genomes project |
| ESP6500 AF (average) | Average population allele frequency from Exome Sequencing Project’s ESP6500 |
| ESP6500 AF (European American) | Population-specific AF |
| ESP6500 AF (African American) | Population-specific AF |
| gnomAD total AF | Total healthy population allele frequency from the Broad Institute gnomAD project |
| gnomAD AF African | African/African American specific AF |
| gnomAD AF American | Admixed American/Latino specific AF |
| gnomAD AF Ashkenazi Jewish | Ashkenazi Jewish specific AF |
| gnomAD AF East Asian | East Asian specific AF |
| gnomAD AF Finnish | Finnish specific AF |
| gnomAD AF Non-Finnish European | Non-Finnish European specific AF |
| gnomAD AF South Asian | South Asian specific AF |
| gnomAD AF Other | Other population specific AF |
| COSMIC variant count | Total of alt base(s) previously observed in COSMIC database |
| COSMIC variant count (tissue) | Alt base(s) previously observed in COSMIC database, grouped by tissue |
| COSMIC transcript | Transcript used by COSMIC curators |
| COSMIC protein change | Protein sequence change used by COSMIC curators |
| Number of samples with variant | Alt base recurrence in submitted samples |
| Protein 3D Variant | If the variant is mapped to a 3D protein structure, this link opens an interactive visualization window from MuPIT. |
| ClinVar disease identifier Identifier | ClinVar disease identifier |
| ClinVar XRef | Cross-references for ClinVar annotations |
| HGVS Genomic | Human Genome Variation Society genomic nomenclature of variant |
| HGVS Protein | Human Genome Variation Society protein nomenclature of variant in the most damaging S.O. transcript |
| HGVS Protein All | Human Genome Variation Society protein nomenclature for all transcripts |
| HUGO Symbol | Gene symbol from HUGO in which the mutation resides |
| TARGET | Drugs that target the gene from TARGET database |
| CGL Driver Class | Oncogene or Tumor suppressor gene annotated by Cancer Gene Landscapes |
| Number of samples with gene mutated | Gene mutation recurrence in submitted samples |
| CGC driver class | Oncogene or Tumor suppressor gene annotated by Cancer Gene Census |
| CGC inheritance | Somatic or germline annotated by Cancer Gene Census |
| CGC tumor types somatic | Annotations from Cancer Gene Census tumor types somatic |
| CGC tumor types germline | Annotations from Cancer Gene Census tumor types germline |
| COSMIC gene count | Total times gene is mutated in COSMIC (add link) database |
| COSMIC gene count (tissue) | Total times gene is mutated in COSMIC (add link) database, grouped by tissue |
| Protein 3D Gene | If variants in the gene are mapped to a 3D protein structure, this link opens an interactive visualization window from MuPIT. |
| NCI Pathway Hits | Count of genes from the National Cancer Institute Pathway Interaction Database that contain the mutated gene. From Network Data Exchange (NDEX) enrichment service. |
| NCI Pathway IDs | Network Data Exchange (NDEX) identifiers of pathways from the National Cancer Institute Pathway Interaction Database that contain the mutated gene. |
| NCI Pathway Names | Names of pathways from the National Cancer Institute Pathway Interaction Database that contain the mutated gene. From Network Data Exchange (NDEX) enrichment service. |
| UTR/Intron | Mapping to noncoding regions (UTRs, 2k upsteam/downsteam regions, and introns). From UCSC GRCh38 database. |
| UTR/Intron Gene | Gene in which a non-coding variant occurred |
| UTR/Intron All Transcript | Mapping to noncoding regions for each transcript |
| ncRNA Class | Noncoding RNA class |
| ncRNA Name | Noncoding RNA name |
| Repeat Class | Repeated sequence class |
| Repeat Family | Repeated sequence family |
| Repeat Name | Repeated sequence name |
| Pseudogene | Pseudogene a variant occurred in |
| Pseudogene Transcript | Transcript of noncoding pseudogene a variant occurred in |
| GWAS NHBLI Key (GRASP) | GRASP-GWAS NHBLI keys. From GRASP project. |
| GWAS PMID (GRASP) | List of PubMed Ids for associated GRASP phenotypes. List order matches GWAS NHBLI Key. |
| GWAS Phenotype (GRASP) | List of phenotypes in GRASP catalogue with associated p-values. List order matches GWAS NHBLI Key. |
CHASM-3.1
| Field |
Description |
| CHASM score | Score for somatic missense variants. Ranges from 0 (likely passenger) to 1 (likely driver). *In the original CHASM paper the values were reversed, with 0 as likely driver and 1 as likely passenger. |
| CHASM P-value | Empirical p-value (probability that passenger variant is misclassified as a driver). |
| CHASM FDR | False discovery rate expected (Benjamini-Hochberg multiple testing correction). |
| CHASM transcript | Transcript used for CHASM score |
| All transcripts CHASM results | List formatted as TranscriptID:ProteinSequenceChange:CHASMscore:CHASMp-value calculated for all transcripts. *= transcript with most severe CHASM score |
| CHASM gene score | Highest CHASM score in the gene (only missense variants are considered) |
| CHASM gene P-value | Composite p-value for non-silent variants in the gene combined with Stouffer’s Z-score method |
| CHASM gene FDR | Composite false discovery rate (Benjamini-Hochberg multiple testing correction) for non-silent variants in the gene combined with Stouffer’s Z-score method. |
VEST-4 Still using VEST-3? Available on
hg19.cravat.us.
| Field |
Description |
| VEST Score (missense) | Pathogenicity score for missense germline variants |
| VEST Score (frameshift indels) | Pathogenicity score for frameshift insertion and deletion germline variants |
| VEST Score (inframe indels) | Pathogenicity score for in-frame insertion and deletion germline variants |
| VEST Score (stop-gain) | Pathogenicity score for stop-gain germline variants |
| VEST Score (stop-loss) | Pathogenicity score for stop-loss germline variants |
| VEST Score (splice site) | Pathogenicity score for splice site germline variants |
| VEST P-value | Empirical p-value (probability that benign variant is misclassified as pathogenic). |
| VEST FDR | False discovery rate expected (Benjamini-Hochberg multiple testing correction). |
| All transcripts VEST results | List formatted as TranscriptID:ProteinSequenceChange:VESTscore:VESTp-value calculated for all transcripts. *= transcript with most severe VEST score. |
| VEST gene score (non-silent) | Highest pathogenicity score in the gene (all non-silent variants are considered) |
| VEST gene P-value | Composite p-value for non-silent variants in the gene combined with Stouffer’s Z-score method |
| VEST gene FDR | Composite false discovery rate (Benjamini-Hochberg multiple testing correction) for non-silent variants in the gene combined with Stouffer’s Z-score method. |
| Field |
Description |
| GeneCards summary | GeneCards annotation |
| PubMed article count | Number of the records retrieved from PubMed, using the name of the gene which contains the mutation and "cancer" as keywords. |
| PubMed search term | PubMed search result link. |